Simple comparison chart for alpha vs. beta thalassemia:
| Feature | Alpha Thalassemia | Beta Thalassemia |
|---|---|---|
| Cause | Missing alpha genes | Faulty beta genes |
| Types | Mild to severe (worst = fatal at birth) | Mild to severe (worst needs lifelong transfusions) |
| Hemoglobin F | ❌ Not much change | ✅ Increases a lot in severe cases |
| Main Problem | Less hemoglobin, but some still works | Body makes no normal hemoglobin |
| Common Where? | Asia, Africa, Mediterranean | Mediterranean, Middle East, South Asia |
| Treatment | Usually mild, but severe cases need transfusions | Severe cases need regular transfusions + iron removal |
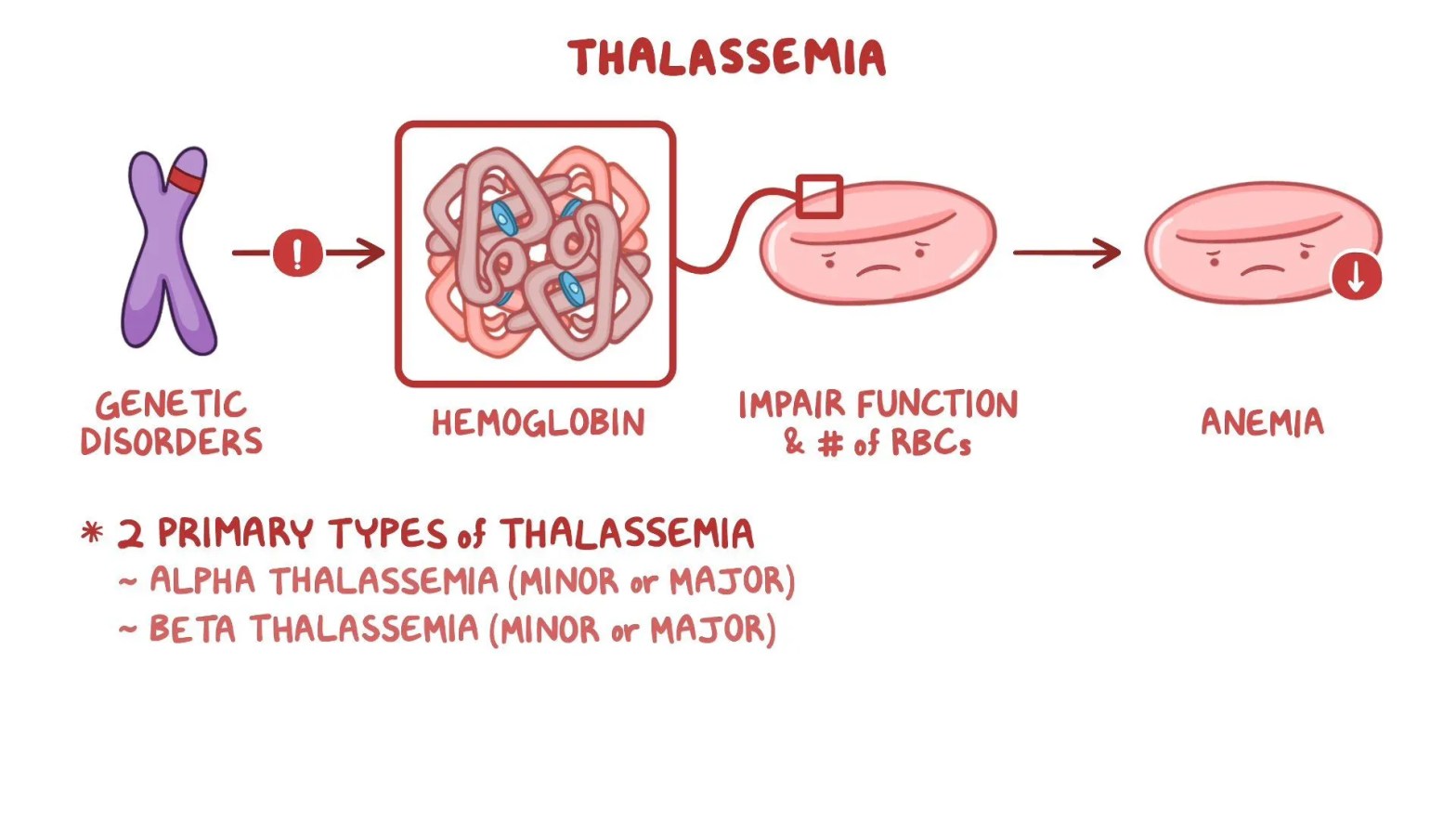
Thalassemia is a blood disorder where the body doesn’t make enough hemoglobin, leading to anemia.
There are two main types:
- Alpha Thalassemia – Problem with alpha hemoglobin.
- Mild (no or few symptoms).
- Moderate (some anemia).
- Severe (can be life-threatening).
- Beta Thalassemia – Problem with beta hemoglobin.
- Mild (mild anemia).
- Moderate (needs occasional treatment).
- Severe (needs regular blood transfusions).
It’s a genetic condition, meaning it’s inherited from parents.
Thalassemia is a genetic blood disorder where the body makes less hemoglobin than normal. Hemoglobin carries oxygen in red blood cells, so a deficiency leads to anemia. There are two main types:
- Alpha Thalassemia – Caused by missing or defective alpha-globin genes.
- Silent Carrier: No symptoms, just one gene missing.
- Alpha Thalassemia Trait (Minor): Mild anemia, two genes missing.
- Hemoglobin H Disease: Moderate to severe anemia, three genes missing.
- Hydrops Fetalis: Severe, often fatal before birth, all four genes missing.
- Beta Thalassemia – Caused by mutations in the beta-globin gene.
- Beta Thalassemia Minor (Trait): Mild anemia, one faulty gene.
- Beta Thalassemia Intermedia: Moderate anemia, two faulty genes but still some hemoglobin production.
- Beta Thalassemia Major (Cooley’s Anemia): Severe anemia requiring lifelong blood transfusions, both genes missing or severely mutated.
Beta thalassemia major (Cooley’s anemia) has increased hemoglobin F (HbF).
Why?
- Beta thalassemia is caused by mutations in the beta-globin genes, leading to little or no production of beta chains of hemoglobin.
- Normal adult hemoglobin (HbA, made of α2β2) cannot form properly due to the lack of beta chains.
- To compensate, the body increases production of fetal hemoglobin (HbF), which is made of α2γ2 (alpha and gamma chains).
- HbF is usually replaced by adult hemoglobin after birth, but in beta thalassemia, it remains high because the body has no functioning beta chains for normal hemoglobin production.
Here’s a simple comparison chart for alpha and beta thalassemia:
| Feature | Alpha Thalassemia | Beta Thalassemia |
|---|---|---|
| Cause | Deletion of alpha-globin genes | Mutation in beta-globin genes |
| Types | – Silent Carrier (1 gene missing) – Alpha Thalassemia Trait (2 genes missing) – Hemoglobin H Disease (3 genes missing) – Hydrops Fetalis (4 genes missing, fatal) | – Beta Thalassemia Minor (1 gene affected, mild anemia) – Beta Thalassemia Intermedia (moderate anemia) – Beta Thalassemia Major (Cooley’s anemia, severe anemia) |
| Hemoglobin F (HbF) Increase? | ❌ No significant increase (except in severe cases) | ✅ Yes, especially in Beta Thalassemia Major |
| Hemoglobin Electrophoresis Findings | Normal or ↓ HbA, HbH present in Hemoglobin H disease | ↑ HbF, ↓ or absent HbA, possible ↑ HbA2 |
| Microcytic Anemia? | ✅ Yes | ✅ Yes |
| Clinical Symptoms | Mild to severe anemia, hepatosplenomegaly in severe cases | Severe anemia, hepatosplenomegaly, skeletal deformities (due to bone marrow expansion) |
| Treatment | Depends on severity; transfusions may be needed for severe cases | Blood transfusions, iron chelation, bone marrow transplant (for severe cases) |
| Geographic Prevalence | Common in Southeast Asia, Africa, and Mediterranean populations | Common in Mediterranean, Middle East, and South Asian populations |
Question:
A 2-year-old boy of Mediterranean descent is brought to the clinic for evaluation of pallor and fatigue. His parents report that he has been lethargic and not eating well. Physical examination reveals hepatosplenomegaly. Laboratory studies show microcytic anemia, low hemoglobin, and increased hemoglobin F on electrophoresis. Which of the following is the most likely diagnosis?
A) Iron deficiency anemia
B) Alpha thalassemia minor
C) Beta thalassemia major
D) Lead poisoning
E) Sickle cell disease
Answer: C) Beta thalassemia major
Explanation:
- The child has microcytic anemia (small red blood cells) and increased hemoglobin F, which is a key finding in beta thalassemia major.
- Mediterranean descent is a risk factor.
- Hepatosplenomegaly happens due to excessive red blood cell destruction.
- Iron deficiency anemia (A) would not show increased hemoglobin F.
- Alpha thalassemia minor (B) usually has mild anemia without major symptoms.
- Lead poisoning (D) can cause anemia but would also show neurological symptoms.
- Sickle cell disease (E) presents with pain crises and sickle-shaped cells, not microcytosis.
